Materials and Methods
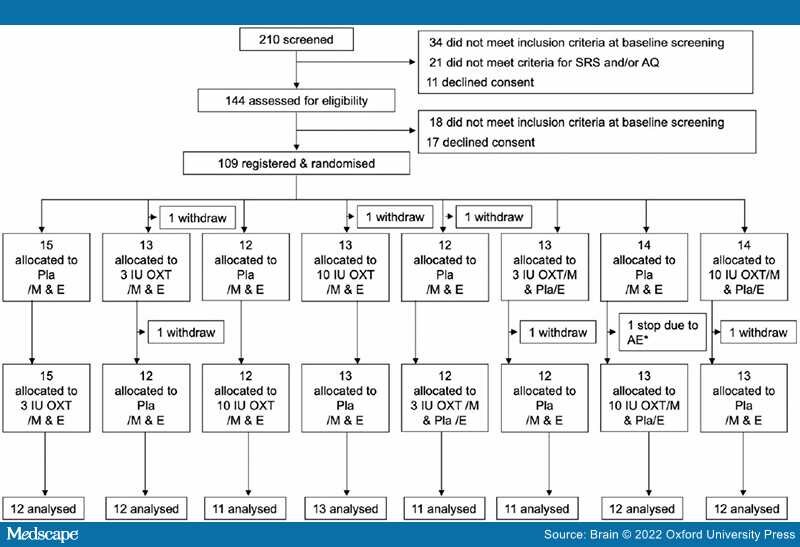
The present double-blind, placebo-controlled, multicentre, crossover trial with two administration periods was conducted at seven university hospitals in Japan. The recruitment process and screening, primary outcome scoring and data and drug management were performed at the main site (Hamamatsu University School of Medicine Hospital). Testing eligibility, confirmation of diagnosis, registration, oxytocin/placebo treatments and clinical assessments were performed at each site (further details and the and prespecified full protocol are provided in the Supplementary material). The study protocol was approved by the institutional review boards for each site and was registered (ClinicalTrials.gov Identifiers: NCT03466671/UMIN000031412). Written informed consent was obtained from all participants at each site.
Eligibility Criteria for Participants
Inclusion criteria were as follows: (i) males aged 18–55; (ii) ASD diagnosis based on the Diagnostic and Statistical Manual of Mental Disorders-5 (DSM-5)[36] with a score exceeding the cut-off value of 10 for qualitative abnormalities in social reciprocity on the Autism Diagnostic Interview Revised;[37] and (iii) Full Scale Intelligence Quotient above 80, measured using the Wechsler Adult Intelligence Scale-III.[38]
Exclusion criteria were as follows: (ii) diagnosis of bipolar disorder or schizophrenia spectrum disorder; (ii) primary diagnosis of psychiatric disorders other than ASD; (iii) instability in symptoms of comorbid mental disorders; (iv) history of changes in medication or doses of psychotropics within 1 month before registration; (v) current treatment with more than one psychotropic; (vi) history of hypersensitivity to oxytocin; (vii) history of seizures or traumatic brain injury with loss of consciousness for longer than 5 min; (viii) history of alcohol-related disorders, substance abuse or addiction; (ix) family history of male breast cancer; and (x) severe complications or known hypersensitivity to some drugs and foods.