This transcript has been edited for clarity.
Martha Grogan, MD: Hello, and welcome back to the Mayo Clinic–Medscape Cardiology video series. I'm Martha Grogan. I am the director of the Cardiac Amyloid Clinic at Mayo Clinic in Rochester, Minnesota.
Today I'm really thrilled to have my colleagues with me, Dr Julie Rosenthal, who is the director of the Cardiac Amyloid Program at Mayo Clinic in Arizona, and Dr Melissa Lyle, who is the cardiac amyloid lead for the Multidisciplinary Amyloid Clinic at Mayo Clinic in Florida. Welcome, Julie and Melissa.
We are going to focus on the new transthyretin amyloidosis (ATTR) therapies today, but I am just going to briefly mention that cardiologists should know that amyloid light-chain amyloidosis (AL) treatment is really improving, especially with the use of daratumumab. This is really a game changer, and with all types of amyloidosis we are now seeing improved outcomes.
We are going to briefly talk a little bit about what is transthyretin amyloid and what are the drugs that we have to treat this condition. Transthyretin is a protein that we all make; it is produced in the liver, and it transports thyroid hormone and retinol binding protein throughout the body. Here, one sees the structure of the TTR protein. Remember that there are two main types of amyloidosis that can infiltrate the heart: AL, which is due to a monoclonal disorder of the bone marrow, and transthyretin (TTR)-type amyloid, where the protein is produced in the liver.
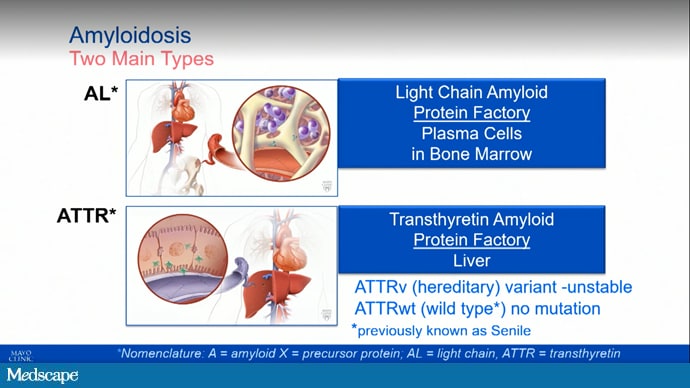
Of the ATTR types, there are both wild-type (which is much more common) and hereditary form. Just a quick overview of that.
When we look at TTR again, and TTR amyloid, there is no actual disease of the liver. The problem is after the protein is formed. The liver produces TTR, and it should stay intact, but in ATTR, it breaks apart. Those monomers then kind of glom together and form amyloid that infiltrates the heart, nerves, and other organs and tissues in the body.
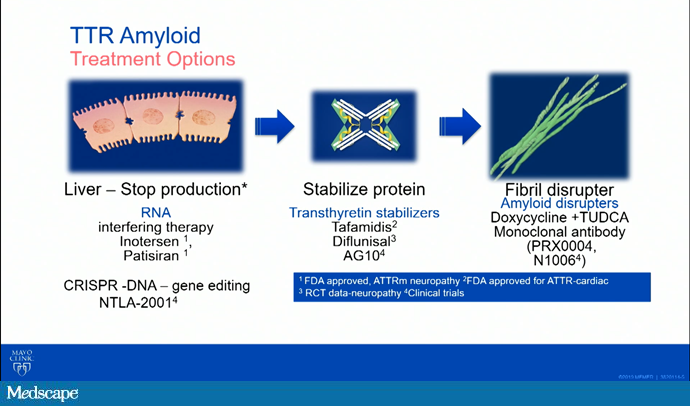
As we talk about these drugs, the reason I showed you that movie is we have a site of action being at the liver, we can have stabilizer drugs that happen at the level of the actual protein, and we can talk about fibril disrupters or degraders. Here, I have listed some of the names of the medications, whether they are in clinical trials, or whether they are approved for use.
Now we are going to go on and talk a little bit about these therapeutics. I am going to start with Melissa. Could you talk to us about the TTR stabilizers?
Melissa A. Lyle, MD: Absolutely. The first one I want to mention is tafamidis. Tafamidis is a once-a-day oral medication that stabilizes the TTR tetramer by binding to the T4 binding site. Actually, this was the first therapy for ATTR cardiac amyloidosis that was approved by the FDA, back in May 2019. That was based on the ATTR-ACT trial.
The ATTR-ACT trial was a randomized, double-blinded study that enrolled 441 patients to either 80 or 20 mg of tafamidis or placebo. The trial demonstrated a 30% reduction in all-cause mortality with tafamidis compared with placebo, as well as a 32% reduction in cardiovascular hospitalizations in patients who were New York Heart Association (NYHA) Class I or II.
Some of the secondary endpoints from that trial really demonstrated a reduction in the decline in the 6-minute walk and also a lower rate in the decline of quality of life, which was assessed by the Kansas City Cardiomyopathy Questionnaire (KCCQ). Subsequent analysis showed that there was a further reduction in all-cause mortality with the 80-mg dose, which is why that is the preferred dose. Tafamidis is really our FDA-approved therapy for ATTR cardiac amyloidosis.
Grogan: Great. A question that comes up all the time is once a patient is on tafamidis, how do we follow them? Any guidelines for our audience in that regard?
Lyle: There is no great way currently. The drug works by slowing the progression of disease. It doesn't necessarily reverse the disease. If there is any sort of clinically relevant regression, that would take years to decades to show. The ways that we follow patients are mostly by clinical assessment. We continue to follow them clinically by physical exam and their general heart failure symptoms.
We also found that frequent serial echocardiogram imaging is not that useful. Follow-up echocardiograms can be useful if there has been progression of symptoms and we think that the echo might change our management, or if they are getting serial echoes for another reason — for example, concomitant aortic stenosis. Otherwise, we are not getting frequent serial imaging.
Serum TTR levels are an indirect measure of stabilization. It is very reasonable to get a serum TTR level at baseline and once after therapy has been started. The utility of serial serum TTR levels has yet to be determined because currently there is no role for a change in dose or uptitration of medication, but that might potentially play a role as a biomarker in the future. Right now, it is mostly following the patient clinically.
Grogan: That is great. Again, we know that there really is no need for safety monitoring. Sometimes people think there are safety labs that we need, but this is a very well-tolerated drug.
I might briefly mention that diflunisal is also an excellent stabilizer. It has not been studied in a randomized trial for cardiomyopathy specifically, but for patients who have adequate renal function, sometimes we need to use diflunisal because of the financial toxicity of tafamidis. How about anything else on the horizon for stabilizers?
Lyle: Yes. That is a great point about the diflunisal. We just caution about monitoring renal function as well as worsening heart failure symptoms.
In terms of other potential options in the future, AG10 is another stabilizer and it actually mimics the stabilizing capabilities of the TTR variant T139M. This is a variant that has been shown in the past to actually be protective against development of polyneuropathy in patients that are heterozygous for the V50M mutation.
This is another stabilizer where phase 2 studies have shown some degree of increase in serum TTR levels as well as stabilization of the TTR tetramer, and phase 3 studies are currently ongoing. Hopefully, we will have new information soon about additional stabilizers.
Grogan: Great. Thanks so much. That was at the level of the protein. Julie, how about if we go back a step to the liver? Tell us about the silencer therapies that we now have available for ATTR.
Julie L. Rosenthal, MD: Thank you. We currently have two silencer therapies approved specifically for individuals living with hereditary ATTR (hATTR), the polyneuropathy phenotype. These two silencers, though having different mechanisms, ultimately have the same goal, and that goal is to prevent translation of that TTR protein so that we have a decrease in the serum concentration of transthyretin.
As I mentioned, they are mechanistically a little bit different. The first type of silencer we have is called patisiran. This was first approved back in 2018, specifically in hATTR patients with polyneuropathy. The clinical trial, APOLLO, demonstrated an overall improvement in these patients' polyneuropathy score, which is four parts of nature, looking at autonomic dysfunction, sensory loss, strength, and reflexibility. It showed improvement over time, suggesting that the silencer not only halts the progression of amyloidosis but that perhaps there is also a potential for regression.
We also saw in this clinical trial an improvement in overall quality of life in these patients receiving silencer therapy. Patisiran specifically is an infusion medication that is received every 3 weeks by these patients. Just like we heard with our stabilizer therapies, these are lifelong therapies, specifically the silencer, meaning they are not there to completely erase the disease. They are there to slow or delay the progression of disease, but it is going to require lifelong therapy.
In order to receive the infusion, the patients do require premedication therapies, specifically with steroids, as well as an antipyretic and antihistamine. That means every 3 months they will be receiving dexamethasone to prevent any sort of infusion reaction.
In addition to patisiran, we have another silencer, called inotersen. Inotersen, like patisiran, halts the progression of our TTR protein formation. Like the APOLLO trial, we had NEURO-TTR, which specifically looked at patients with hereditary polyneuropathy. Like in the APOLLO-B trial, we also saw an improvement in their overall neuropathy function, as well as quality of life.
Similar to patisiran, this too will be a lifelong therapy, but the administration of inotersen is something patients can do in the comfort of their own home by providing themselves with a subcutaneous injection once a week. However, this medication is a little bit different, and it does require specific monitoring, as we did see some safety signals particular with regard to thrombocytopenia and glomerulonephritis. Renal function and platelet counts do need to be followed.
Grogan: Would you tell the audience what happens to the serum TTR levels on the silencer therapy?
Rosenthal: Excellent question. Unlike stabilizers, where we see an increase in our TTR concentration — and if you are looking in your own labs, that would be mimicked as your prealbumin levels — in individuals with silencers. Because we are preventing formation of TTR, you will see an overall decrease in your prealbumin levels.
Grogan: I think that is a really important clinical point. Again, the silencers right now are only approved for hATTR patients with neuropathy. They can have cardiac involvement but not isolated cardiac involvement. Then there are what I call the next generation of both of these drugs, which are easier to administer, subcutaneous, sometimes once a month or every 3 months, and maybe even less than that.
There is much going on with cardiac trials now, both with these agents but also with the next generation. We are really going to have some exciting data coming in the future on how to use these for our patients with amyloid cardiomyopathy.
Melissa, can you tell us what is going on as far as disrupting the fibrils?
Lyle: There are several emerging agents for disruption or degradation. There are two phase 1 studies. The first is for PRX004, an intravenous monoclonal antibody administered via IV infusion every 28 days, and it clears amyloid deposits by binding to the misfolded TTR.
Another form is N1006, which is a recombinant IgG1 human monoclonal antibody. This really is aimed at targeting the misfolded and aggregated form of TTR. Of course, they're just phase 1 studies currently, but many new and exciting things are on the horizon for degradation and disruption of those amyloid fibrils.
Grogan: That is always something that patients ask about. I will comment that there has been much interest in this type of therapy, including in AL. There probably will be some newer trials going on, but it is not an easy thing anyway. I have always wondered: If you actively degrade the fibrils, what will be left behind? We have a lot to learn in this area.
Some providers might see patients who are on the combination of doxycycline and TUDCA (doxycycline-tauroursodeoxycholic acid), which was promoted based on some animal studies, as far as being a fibral disruptor. You might still see patients on that. If they are doing really well, sometimes we continue them, but we are not usually initiating patients on that therapy at this time.
Julie, back to the liver — you are the liver expert. We have heard a lot in the news about CRISPR therapy, particularly for ATTR.
Rosenthal: CRISPR/Cas9 intervention is a gene-suppression therapy that made headlines this past June in The New England Journal of Medicine, specifically looking at only six patients with hereditary polyneuropathy. This was a phase 1 clinical trial. I think it is far from prime time but certainly exciting. With the use of CRISPR therapy, what we saw we could do is induce the cells' natural repair process to overall delete that TTR mutation and knock down all TTR formation.
The question is, will this actually work and clinically have benefits? I think there is more to come, but it's certainly exciting, particularly if we think about our other current therapies that are requiring administration daily, weekly, or every few weeks. The idea of this is it is just a one-time infusion. Certainly, more to come.
Grogan: It is really amazing when we went from almost no treatment for this disease — well, nothing for wild-type ATTR; other than organ transplant, liver transplant was our only real therapy for hATTR, to all of these potential therapies. It is just an explosion in a good way.
I want to highlight for the audience that sometimes the most important thing we can do for our patients is to give them resources: where to get more education and also stay informed about clinical trials. We have a Mayo Clinic Amyloidosis YouTube channel, believe it or not. There are many excellent patient support groups and patient advocacy groups, including the Amyloidosis Foundation, Amyloidosis Research Consortium, and the Amyloidosis Support Groups. Also, for faculty members — it's not listed here — there is a group that really helps bring education to medical students. It can help you with your faculty if you need to make sure that medical students are learning about amyloidosis, and of course, the three of us think there's need for more education.
I'd just highlight all the different things that are on our YouTube channel, which includes a genetic counselor. Of course, that is not a substitute for a genetic counselor in person, but there is some really important information for our patients on those YouTube videos that are available.
It has been our pleasure to share this information with you. Thank you so much to Julie and Melissa. Thank you to our audience for joining us on theheart.org | Medscape Cardiology.
Follow theheart.org | Medscape Cardiology on Twitter
Follow Medscape on Facebook, Twitter, Instagram, and YouTube
Credits
Image 1: Mayo Clinic
Image 2: Mayo Clinic
© 2021 Mayo Clinic
Cite this: Transthyretin Amyloid and New Treatment Options - Medscape - Dec 06, 2021.










Comments