This article originally appeared on the Hospital for Special Surgery's website.
Case Report
A 45-year-old woman with a history of systemic lupus erythematosus (SLE) since 2006 came to the Hospital for Special Surgery in 2014 for continued management of SLE. Disease manifestations included arthritis, alopecia, Raynaud phenomenon, lymphadenopathy, pericarditis, and mononeuritis multiplex. She also had a recurrent erythematous, tender rash on her lower legs; biopsy revealed leukocytoclastic vasculitis with deposits of immunoglobulin M (IgM) and to a lesser extent IgA and C4d within the microvasculature, consistent with an SLE-associated immune complex–mediated vasculitis.
Positive serology results included antinuclear antibody, anti-Smith, anti-Ro, anti-La, and anti-RNP antibodies; low levels of serum complement C3 and C4 were found. Results were negative for anti-dsDNA antibodies, antiphospholipid antibodies, antineutrophil cytoplasmic antibodies, rheumatoid factor, and cryoglobulins.
She was treated with corticosteroids, hydroxychloroquine, azathioprine, tacrolimus, and belimumab. Severe flares of the disease required high-dose corticosteroid therapy.
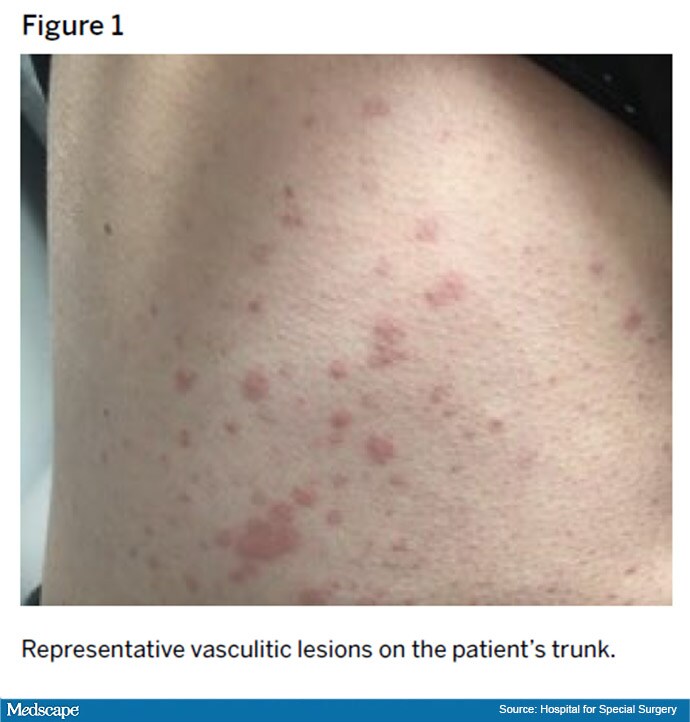
Despite this treatment, she developed progressive disease in 2018, with palpable purpura extending to her proximal legs, arms, and trunk (Figure 1). In the following months, she developed new nephrotic-range proteinuria and microscopic hematuria. A kidney biopsy showed diffuse class IV glomerulonephritis with prominent intracapillary hyaline thrombi and IgM and IgG mesangial and subendothelial deposits.











